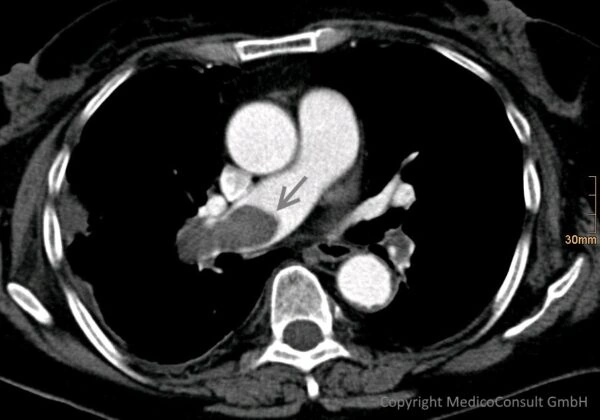
Das Wichtigste
Pulmonale Hypertonie (engl.: pulmonary arterial hypertension, PAH) bedeutet Bluthochdruck in der Lungenarterie, d. h. der Arterie, die vom rechten Herzen zur Lunge führt. Sie ist dadurch gekennzeichnet, dass der Mitteldruck über 25 mm Hg bzw. der systolische Druck über 30 mm Hg (in Ruhe) liegt.
Ursachen: Sie liegen grundsätzlich
in der Lunge, z. B.
oder im linken Herzen, z. B.
- Herzmuskelinsuffizienz im linken Herzen,
- Herzklappenerkrankung.
jeweils mit Rückstau des Bluts in die Lungen und darüber hinaus ins rechte Herz.
Seltenere Ursachen betreffen Septumdefekte zwischen den Vorhöfen oder den Kammern des Herzens mit Übertritt von Blut vom linken ins rechte Herz (Links-Rechts-Shunt).
Die Auswirkungen betreffen eine Blutstauung vor dem Herzen über de Leber (Stauungsleber) bis in den Magendarmtrakt und die Milz und bis in die Beine und in den Kopf mit Funktionsstörungen der betroffenen Organe und einer unzuverlässigen Medikamentenwirkung.
Die Behandlung berücksichtigt vor allem die Ursache. Zur Senkung einer pulmonal-arteriellen Hypertonie kann Bosentan infrage kommen.
Einteilung
Die Einteilung ( 1998 Evian, 2003 Venedig) berücksichtigt die Genese:
- pulmonal-arterielle Hypertonie
- pulmonal-venöse Hypertonie
- pulmonale Hypertonie bei Erkrankungen des Atmungssystems
- pulmonale Hypertonie infolge von Lungenembolien oder chronischer Gefäßprozesse
- pulmonale Hypertonie infolge anderer Erkrankungen, die zu einer Mitbeteiligung der Lungengefäße führen
Formen
In der klinischen Praxis wird die pulmonale Hypertonie anhand der Entstehungsmechanismen in fünf Gruppen eingeteilt:
GRUPPE 1 Pulmonale arterielle Hypertonie (PAH);
GRUPPE 2 PH im Zusammenhang mit einer Erkrankung des linken Herzens;
GRUPPE 3 PH im Zusammenhang mit Lungenerkrankungen und/oder Hypoxie;
GRUPPE 4 PH im Zusammenhang mit Lungenarterienobstruktionen (einschließlich chronische thromboembolische pulmonale Hypertonie, CTEPH);
GRUPPE 5 PH mit unklaren und/oder multifaktoriellen Mechanismen. 1
Entstehung
Folgende pathophysiologische Überlegungen tragen zum Verständnis einer pulmonalen Hypertonie bei:
- Die präkapillären Blutgefäße sind nur gering muskularisiert und bieten daher dem Blut wenig Widerstand. Der „transpulmonale Gradient“ (TPG) beträgt nur wenige Millimeter. Eine starke Flusserhöhung bei körperlicher Anstrengung kann normalerweise vom Flussbett problemlos bewältigt werden.
- Bei Hypoxie kommt es zu einer Vasokonstriktion (Euler-Liljestrand-Reflex), während im großen Kreislauf unter solchen Bedingungen eine Vasodilatation folgt. Durch die Vasokonstriktion werden nicht durchblutete Lungenbezirke aus der Blutbahn ausgeschaltet, was bewirkt, das nicht oxigeniertes Blut das oxigenierte nicht verdünnt. Die Oxigenierung des arteriellen Bluts im großen Kreislauf sinkt damit auch bei Ausschaltung von Lungenbezirken aus der Ventilation kaum.
- Bei der primären pulmonalen Hypertonie ist eine pulmonal-endotheliale Dysfunktion mit konsekutiver Vasokonstriktion von zentraler Bedeutung. Es kommt zu pathologischen vaskulären Proliferationen (pulmonal-vaskuläres Remodeling) und Mikrothrombenbildungen. Letztlich entstehen Intimafibrose und Verdickung der Media, die eine Widerstandserhöhung der pulmonalen Gefäße bewirken. 2
- Bei der ausgeprägten chronischen Linksherzinsuffizienz mit konsekutiver Mitralklappeninsuffizienz kommt es ebenfalls zu einer pulmonal-endothelialen Dysfunktion mit letztlich einer Erhöhung des Gefäßwiderstands (pathophysiologische Endstrecke wie oben).
- Die rechtsventrikuläre Ejektionsfraktion bestimmt bei Widerstandserhöhung im kleinen Kreislauf wesentlich die Belastbarkeit und Prognose. Ist die rechte Kammer schwach (z. B. bei Myokarditis oder koronarer Ischämie), kommt es unter Belastung zu einem Abfall des Füllungsdrucks im linken Vorhof; die linkskardiale Ejektion sinkt.
- Bei rechtsventrikulärer Dekompensation kommt es zur Gefügedilatation des Ventrikels, wodurch sich auch der Klappenring der Trikuspidalklappe weitet. So kommt es zu ihrer Schlussinsuffizienz (Trikuspidalinsuffizienz). Dies hat zur Folge, dass der Druck in der Pulmonalarterie nicht weiter ansteigt und ein positiver Jugularvenenpuls klinisch sichtbar wird.
- Das Schlafapnoe-Syndrom wird als wichtiger Faktor, der eine pulmonale Hypertonie fördern kann, diskutiert. 3
- Der Links-rechts-Shunt bei einem Vorhofseptumdefekt oder einem Ventrikelseptumdefekt führt zu einer Volumenbelastung des rechten Herzens und der kleinen Kreislaufs mit Ausbildung einer pulmonalen Hypertonie.
Exosomen in der PAH-Entstehung
- Extrazelluläre Vesikel (Exosomen, EV) sind durch die Expression von Tetraspaninen charakterisiert und übertragen Moleküle, die zum Fortschreiten der PAH beitragen, so insbesondere Chemokine, Zytokine, Lipide, RNA und miRNA. Bei einer pulmonalen Hypertonie wird eine erhöhten EV-Zahl festgestellt, die von Immunzellen (T-Lymphozyten) stammen. Daher wird angenommen, dass die PAH eine „entzündliche Erkrankung“ darstellt, die zu einer Ummodellierung der kleinsten Blutgefäße in den Lungen führt. 4 EVs korrelieren mit dem mittleren Lungenarteriendruck. 5 Es wurden auch aus Blutplättchen stammende EVs bei pulmonaler Hypertonie gefunden, die prothrombotische Eigenschaften aufweisen und die pulmonale Hypertonie verstärken können. 6
Erbliche PAH
Die vererbbare PAH wird durch einen genetischen Funktionsverlust im Protein (BMP)-Rezeptor 2 (BMPR2) verursacht; dies ist ein Signalrezeptor der transformierenden Wachstumsfaktor-β (TGF-β)-Superfamilie. Es kommt zu einer unausgewogenen Signalübertragung der TGF-β-Superfamilie: einer verringerten Signalübertragung über BMP-Liganden und einer erhöhten Signalübertragung über TGF-β-Liganden. Bisherige Behandlungen basierten auf Vasodilatatoren, die den zugrunde liegenden Krankheitsmechanismus jedoch nicht korrigieren. Es werden Therapien erforscht, die direkt auf die dysregulierten Signalwege abzielen 7.
Diagnostik
Anamnese: Hinweise auf Thrombosen oder Lungenembolien? Vorbekannte Linksherzinsuffizienz? Koronare Herzkrankheit?
Klinisches Bild: Atemnot, gestaute Halsvenen, hepatojugulärer Reflux bei Druck auf die Leber. Ggf. Zyanose, Lungenemphysem, Zeichen abgelaufener Thrombosen an den Beinen. Mitralinsuffizienzgeräusch. Untersuchung auf Zeichen einer Leberstauung bzw. einer Cirrhose cardiaque.
EKG: P pulmonale, Linksverschiebung der Umschlagszone, u. U. Rechtsschenkelblock
Echokardiographie: Abschätzung des pulmonal-arteriellen Drucks und der rechtskardialen Ventrikelfunktion
- Schweregrad leicht – NYHA I – PAP 35-55 mm Hg
- Schweregrad mäßig – NYHA II – PAP > 55 mm Hg
- Schweregrad schwer – NYHA III – reduzierte rechtsventrikuläre Funktion
- Schweregrad sehr schwer – NYHA IV – stark reduzierte rechtsventrikuläre Funktion
Rechtsherzkatheter: Abschätzung des pulmonal-arteriellen Drucks und der rechtskardialen gemischtvenösen Sauerstoffsättigung
- Schweregrad leicht – NYHA I – PAM 21-40 mm Hg
- Schweregrad mäßig – NYHA II – PAM > 40 mm Hg
- Schweregrad schwer – NYHA III – SvO2 < 60%
- Schweregrad sehr schwer – NYHA IV – SvO2 < 50% (vitale Bedrohung)
Schlaflabor zur Diagnostik eines Schlafapnoe-Syndroms (wenn die Anamnese dazu Veranlassung gibt)
Weitere Diagnostik: Die Diagnostik beinhaltet die Suche nach der Ursache der pulmonalen Hypertonie, wie einer Lungenembolie oder einer chronischen Lungen- oder Herzkrankheit, und nach den Folgen, wie einer Rechtsherzinsuffizienz, einer Stauungsleber und einer Blutstauung im Magendarmkanal und in abhängigen Körperpartien.
Therapie
- Medikamentös bei pulmonal-venöser Hypertonie (Lungenhochdruck bei Linksherzversagen): Diuretika und ACE-Hemmer zur Nachlastsenkung bei linksventrikulärer Dekompensation. Zu beachten ist, dass bei erheblicher rechtskardialer Stauung eine portale Hypertension auftreten kann mit negativen Folgen für die orale Bioverfügbarkeit (Resorption) von Medikamenten.
- Medikamentös bei pulmonal-arterieller Hypertonie (Lungenarterienhochdruck wegen einer Lungenkrankheit): Bosentan (Tracleer®, nichtselektiver Endothelin-Rezeptorantagonist), Sitaxentan (Thelin®) und Ambrisentan (Volibris®) (selektive Endothelin-Rezeptorantagonisten). Mehr dazu siehe hier.
- Operative Maßnahmen zur Verbesserung der Koronarperfusion und zur Schlussfähigkeit der Klappen.
- Lungentransplantation in geeigneten Fällen 8.
- Eine ursächliche Therapie ist in einigen Fällen möglich und anzustreben, so die Auflösung eines Embolus (Lysetherapie) im Lungenkreislauf oder ein Klappen- oder Shuntvitium des Herzens. Andere ursächliche Krankheiten, wie eine autoimmune Lungenkrankheit, können medikamentös aufgehalten werden.
- Ein neues therapeutisches Ziel besteht im Angriff auf die mit den vermehrt auftretenden Exosomen transportierten mikroRNA (miR). Erste tierexperimentelle Untersuchungen sind erfolgversprechend. 1
- Rote-Beete-Saft ist nitratreich und führt zu einer Erhöhung der NO-Produktion im Lungenkreislauf (NO erweitert die Blutgefäße). Er kann den pulmonalarteriellen Druck senken und als Begleitbehandlung bei pulmonaler Hypertonie infrage kommen 9.
- Sotatercept verlangsamt die Progression einer pulmonalen Hypertonie in den Stadien III und IV. Die Zulassungsstudie über 1 Jahr wurde wegen der guten Wirkung vorzeitig abgebrochen. Sie zeigte, dass Tod jeglicher Ursache bei 8,1 % in der Sotatercept-Gruppe (vs. 15,1 % in der Placebogruppe) auftrat; eine Lungentransplantation war bei 1 Patienten (1,2 % vs. 7,0 %) erforderlich geworden; ein Krankenhausaufenthalt war seltener notwendig (9,3 % vs. 50,0 %). Die häufigsten unerwünschten Ereignisse mit Sotatercept waren Epistaxis und Teleangiektasien. 10 Erklärung: Sotatercept ist ein Fusionsprotein, das Aktivine und Wachstumsdifferenzierungsfaktoren abfängt, die an der pulmonalen arteriellen Hypertonie beteiligt sind. Laut einer ersten größeren Studie ermöglichte es nach 24 Wochen eine Verlängerung des Gehwegs um durchschnittlich 40 Meter 11.
→ Auf facebook informieren wir Sie über Neues und Interessantes!
→ Verwalten Sie Ihre Laborwerte mit der Labor-App Blutwerte PRO – mit Lexikonfunktion.
Verweise
Weiteres
- Biology (Basel). 2023 Aug 7;12(8):1099. doi: 10.3390/biology12081099[↩][↩]
- Am. J. Respir. Crit. Care Med. 2021;203:1472–1487[↩]
- Proc Am Thorac Soc. 2008 Feb;5(2):200-6[↩]
- Pulm. Circ. 2019;9:1–4. doi: 10.1177/2045894019864357[↩]
- J. Heart Lung Transplant. 2009;28:1081–1086[↩]
- Thromb. Res. 2020;195:120–124[↩]
- Am J Respir Crit Care Med. 2016 Nov 1;194(9):1047-1049. doi: 10.1164/rccm.201605-0920ED[↩]
- Int J Clin Pract Suppl. 2007 Dec;(158):4-9[↩]
- J Card Fail. 2018 Oct;24(10):640-653[↩]
- N Engl J Med. 2025 May 29;392(20):1987-2000. doi: 10.1056/NEJMoa2415160[↩]
- N Engl J Med. 2023 Apr 20;388(16):1478-1490. doi: 10.1056/NEJMoa2213558[↩]