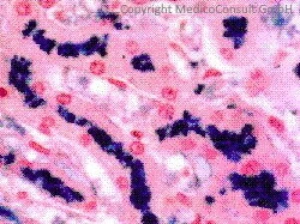
Die Entstehung der Hämochromatose ist weitgehend aufgeklärt. Die idiopathische HFE-Hämochromatose ist eine autosomal-rezessiv vererbte Erkrankung.
Allgemeines
Die Krankheit beruht auf einer Homozygotie für eine Mutation, die zu einer C282Y-Substitution im HFE-Protein führt. Folge ist eine verminderte Expression des eisenregulierenden Hormons Hepcidin. Klinisch folgen eine erhöhte Eisenaufnahme aus der Nahrung und Eisenablagerungen in verschiedenen Geweben wie Leber, Bauchspeicheldrüse, Gelenken, Herz und Hypophyse. Allerdings ist die klinische Präsentation der Eisenüberladung sehr variabel: einige Betroffene zeigen kaum oder gar keine Symptome, andere dagegen schwere Gewebeschäden und klinische Folgen. Dies liegt an verschiedenen begleitenden Umständen, zu denen individuelle genetische Eigenschaften, regelmäßiger Blutverlust (Morbus Osler, Menstruation) oder zusätzlicher Alkoholkonsum gehören können 1.
Genetik
Das 1996 identifizierte Hämochromatose-Gen ist auf Chromosom 6 lokalisiert (HFE-Gen). Typischerweise besteht eine Homozygotie für C282Y oder eine Heterozygotie für C282Y zusammen mit einer Heterozygotie für H63D. Die Compound-Heterozygotie aus einer C282Y- und einer H63D-Mutation liegt bei 2 – 5 % der Hämochromatose-Patienten vor und bewirkt eine mildere Verlaufsform.
C282Y-Mutation: Eine praktisch immer vorliegende C282Y-Mutation beeinträchtigt die Expression des HFE-Proteins an der Oberfläche der Zellmembran der Schleimhautzellen in den Krypten der Duodenalschleimhaut und damit die Aufnahme von Eisen, das durch zirkulierendes Transferrin transportiert wird.
Die C282Y-Mutation im HFE-Gen ist bei Hämochromatose-Patienten keltischer Abstammung häufig zu finden. 2 Sie findet sich nicht oder selten bei Hämochromatose-Patienten nicht-keltischer Abstammung; dort ist von anderen Mutationen auszugehen. Die Mutationen H63D und S65C sind mit einer milderen Verlaufsform assoziiert.
Andere Mutationen führen (seltener) zu einer verstärkten Eisenakkumulation in den Organen oder verstärken sie. Dazu gehören Hämojuvelin (HJV), Transferrinrezeptor 2 (TfR2) und Hepcidin (HAMP). Diese Mutationen findet man vor allem bei Patienten, die eine Hämochromatose bereits im jugendlichen und frühen Erwachsenenalter entwickeln (juvenile Hämochromatose).
Die hereditäre Typ-1-HFE-Hämochromatose ist gekennzeichnet durch das homozygote Vorkommen einer C282Y-Mutation. Sie führt zu einer erniedrigten Bildung von Hepcidin in der Leber. Dieses Protein kontrolliert die Eisenabgabe aus Enterozyten (Zellen der Dünndarmschleimhaut) und aus Makrophagen ins Blut, wo es an Transferrin gebunden zu Organen getragen wird, die es leicht aufnehmen und ansammeln, wie Leber, Herz und Bauchspeicheldrüse. Weitere genetische Faktoren können die Ausprägung der Eisenspeicherung erheblich verstärken.
Eine Homozygotie des H63D-Gens (bis 4 % der Bevölkerung) gilt nicht als disponierend für die Hämochromatose. 3 4 Die Transferrin-Sättigung bei Heterozygoten für H63D ist in der Regel höher als bei normalem Genotyp, wenngleich sie in einer überprüften Population alle im Normbereich lagen. 5 6
Die S65C-Mutation ist – in Kombination mit einer C282Y-Heterozygotie – mit einer milden Verlaufsform assoziiert. 7 Sie prädisponiert ohne zusätzliche Kombination mit einer C282Y- oder H63D-Mutation zu erhöhten Ferritin-Werten. 8
Die H63D- und S65C-Mutationen scheinen die Affinität des Transferrin-Rezeptors zu verändern.
Eine Heterozygotie für die HFE-Genmutationen scheint mit einer Prädisposition für Langlebigkeit assoziiert zu sein. 9
Eine erhöhte parenterale Eisenzufuhr, z. B. durch Transfusionen, führt unter Umgehung der Darmmukosa zur erworbenen Hämochromatose.
Entstehung
Idiopathische Hämochromatose
Die Entstehung der Hämochromatose hängt mit dem Verlust der Dünndarmzellen zusammen, die Eisenaufnahme zu regulieren.
Die Homöostase (Aufrechterhaltung des Gleichgewichts von Aufnahme und Verlust) des Körpereisens wird lediglich durch die Eisenresorption im Dünndarm reguliert. Einen spezifischen Ausscheidungsmechanismus für Eisen gibt es nicht. Bei der Hämochromatose ist die enterale Eisenresorption gesteigert, der Eisentransit durch die Mukosazellen im Darm ist beschleunigt und so die Eisenabgabe aus diesen Zellen ins Blut erhöht. Der normale Eisenverlust, der durch Zellabschilferung (vor allem im Darm) erfolgt (es gibt keinen eigenen Eisenausscheidungsmechanismus), wiegt die vermehrte Aufnahme nicht auf; es kommt zu einer positiven Eisenbilanz. Blutverluste können die klinische Symptomentwicklung hinauszögern (Menstruation bei Frauen). Bei Heterozygoten findet sich häufig eine Hämosiderose (vermehrte Eisenspeicherung, aber nicht so stark, siehe hier) und wohl nur in Ausnahmefällen das Vollbild einer Hämochromatose.
Erworbene Hämochromatose
- Gehäufte Transfusionen v. a. bei aplastischen Anämien oder Myelofibrosen können unter Umgehung der Darmmukosa zur Eisenüberladung der Leber führen.
- Gesteigerter Hämoglobinumsatz bei Thalassaemia major, hereditärer Sphärozytose und sideroblastischer Anämie führen ebenfalls zur hepatischen Eisenspeicherung.
- Die Porphyria cutanea tarda beruht auf einem Enzymdefekt (Defekt der Uroporphyrinogen-Decarboxylase), der eine verminderte Hämsynthese bedingt. Bei gleichzeitig erhöhtem Alkohol-, Medikamenten- oder Eisenkonsum wird die Eisenüberladung manifest.
- Der Mangel an Transferrin führt bei der Atransferrinämie zur Eisenüberladung der Leber und anderer Organe.
- Eine erhöhte enterale Eisenzufuhr kann bei alkoholischer Hepatopathie zur Hämosiderose, in ausgeprägten Fällen sogar zur Hämochromatose der Leber führen. Durch Aderlässe lassen sich die Eisenspeicher jedoch sehr viel rascher als bei einer genetisch bedingten Hämochromatose leeren.
Eine HFE-Genmutation hat Auswirkungen auf den Eisenstatus bei chronischer Hepatitis C, bei der eine oft beobachtbare Eisenakkumulation in der Leber mit einer verstärkten Fibroseneigung und einer schlechteren Therapierbarkeit einhergeht.
Einzelne Faktoren im Eisenstoffwechsel
Mutationen des HFE-Gens, von Hämojuvelin (HJV) und des Transferrinrezeptors 2 (TfR2) führen zu niedrigen Hepcidinspiegeln im Blut. Eine Eisenüberladung des Körpers kann außer durch Mutation dieser Gene durch Mutationen verschiedener weiterer am Eisenstoffwechsel beteiligter Prinzipien zustande kommen 1. Sie führen z. T. zu einer beschleunigten Entstehung einer Hämochromatose, wie es bei der juvenilen Form der Fall ist.
Eine zentrale Rolle spielt die ungebremste Eisenaufnahme in Dünndarm. Ursache ist eine Störung des Zusammenspiels des Eisentransporters Ferroportin, des regulatorischen Hormons Hepcidin und des Hemojuvelins, welches die Hepcidin-Expression reguliert:
Ein Hepcidinmangel führt zu einer Eisenüberladung, ein Hepcidinüberschuss dagegen zu einer Anämie. Hemojuveloin ist ein Korezeptor von BMP, ein Protein, welches die Hepcidin-Expression in Leberzellen hochreguliert; Hämojuvelin verstärkt diesen Prozess. In HFE2–/– -Hepatozyten (Leberzellen, die kein HFE-Gen tragen) ist er vermindert. 10 )
Hepcidin
Hepcidin kommt eine zentrale Rolle in Eisenspeicherkrankheiten zu. Es ist der Hauptregulator des Eisenstoffwechsels und wird von Leberzellen (Hepatozyten) als Reaktion auf eine erhöhte Eisenkonzentration im Körper und auf Entzündungen synthetisiert und freigesetzt. Seine Bildung wird durch Eisen hoch und durch Eisenmangel herunterreguliert 11.
Ein genetischer Defekt der Bildung von Hepcidin führt zur Eisenüberladung des Körpers bereits in der Jugend (juvenile Hämochromatose) 12.
Hepcidin bindet an die Plasmamembranen der Mukosazellen der Dünndarmschleimhaut. Bindungspartner ist Ferroportin, das durch die Bindung in seiner Fähigkeit, Eisen ans Plasma (Transferrin) abzugeben, inaktiviert wird (s .u.). Ein niedriger Hepcidinspiegel bedeutet damit eine hohe Transportaktivität der Plasmamembran von Dünndarmmukosazellen (Enterozyten) für Eisen.
Ferroportin
Ferroportin ist ein Eisentransporter der Plasmamembran, der für die Eisenresorption im Dünndarm und zudem für die Ausschleusung von Eisen aus Körperzellen verantwortlich ist. Im Dünndarm ist es in der basolateralen Plasmamembran lokalisiert. Die experimentelle Inaktivierung von Ferroportin führt zu einer Eisenüberladung von Dünndarmzellen, Hepatozyten und Makrophagen. 13 Eine Mutation von Ferroportin ist Ursache der Hämochromatose Typ IV (HFE4), die sich von der klassischen Hämochromatose durch besonders hohen Eisengehalt der Kupffer-Zellen abhebt. 14
Ferroportin wirkt als Rezeptor für Hepcidin und wird durch die Bindung inaktiviert. Wenn also Hepcidin hochreguliert wird, ist die Transportkapazität an den Dünndarmzellen für Eisen niedrig und umgekehrt. 15
Hemojuvelin
Hemojuvelin wurde in Leber, Herz, Ösophagus, Pankreas, Colon descendens, Coecum, Ileum und Skelettmuskulatur gefunden. 16 In der Leber wird Hemojuvelin in periportalen Hepatozyten gebildet und bewirkt eine Senkung des Hepcidinspiegels im Blut. Eine Mutation von Hemojuvelin bewirkt eine geringere Unterdrückung von Hepcidin und damit eine vermehrte Degradierung von Ferroportin. Dies wird als Ursache einer Hämochromatose (Typ II) angesehen. Die periportalen Zellen übernehmen damit möglicherweise eine entscheidende Funktion im „Eisensensing“, worüber die Hepcidin- und damit die Ferroportinaktivität geregelt wird. 17 Hemojuvelin wird offenbar besonders durch Zytokine reguliert, wodurch es eine Schlüsselfunktion in der entzündungsbedingten „Downregulation“ des Serumeisens zu haben scheint. 18
Transferrinrezeptor2 (TFR2)
Der Transferrinrezeptor2 (TFR2) wird in Hepatozyten und Erythrozyten-Vorläufern gefunden. Er bildet einen Komplex mit dem Hämochromatoseprotein HFE. Eine defekte TFR2-Funktion verursacht eine Herunterregulierung der Hepcidinbildung und darüber eine systemische Eisenüberladung mit Ausbildung einer Hämochromatose 19.
→ Auf facebook informieren wir Sie über Neues und Interessantes!
→ Verwalten Sie Ihre Laborwerte mit der Labor-App Blutwerte PRO – mit Lexikonfunktion.
Verweise
- Hämochromatose Hauptblatt
- Hämochromatose Diagnostik
- Hämochromatose Therapie
- Hepcidin
- Eisenstoffwechsel
- Eisen
- Ferritin
- Transferrin
- Löslicher Transferrinrezeptor
Weiteres
- Ann Transl Med. 2021 Apr;9(8):731. doi: 10.21037/atm-20-5512[↩][↩]
- Nat Genet 1996; 13: 399-408; Blood Cells Mol Dis 1996; 22: 187-194[↩]
- N Engl J Med. 2005 Apr 28;352(17):1741-4.[↩]
- Gastroenterolgy 1999; 116: 193-207[↩]
- Eur J Epidemiol 2003; 18: 685-689[↩]
- Gastroenterology 1999; 116: 1409-1412[↩]
- Blood 1999; 15: 2502-2505[↩]
- Gut 2002; 51: 723-730[↩]
- Mech Ageing Def 2003: 124: 529-532[↩]
- Nat Genet 38, 2006; 531–539. https://doi.org/10.1038/ng1777[↩]
- Vitam Horm. 2019;110:71-99. doi: 10.1016/bs.vh.2019.01.004[↩]
- Best Pract Res Clin Haematol. 2005; 18:171-82[↩]
- Cell Metab. 2005; 1: 191-200[↩]
- Proc Natl Acad Sci 2005; 102: 8955-60; Blood Cells Mol Dis. 2005; 35: 33-46[↩]
- Annu Rev Pathol. 2009;4:489-515[↩]
- Haematologica. 2004; 89: 1441-5[↩]
- J Clin Invest. 2005; 115: 2180-6[↩]
- J Mol Med. 2005; 83: 521-5[↩]
- Free Radic Biol Med. 2019 Mar;133:46-54. doi: 10.1016/j.freeradbiomed.2018.06.037[↩]
- Clin Gastroenterol Hepatol. 2025 Aug;23(9):1477-1485. doi: 10.1016/j.cgh.2024.10.041.[↩]
- J R Coll Physicians Edinb. 2024 Dec;54(4):340-347. doi: 10.1177/14782715241298724[↩]
- Ann Intern Med. 2025 Feb;178(2):ITC17-ITC32. doi: 10.7326/ANNALS-24-03710[↩]