Algemeines
Neuroendokrine Tumore (NET) wurden früher als Karzinoide bezeichnet und unter dem Begriff APUDome zusammengefasst. Ein Teil der Tumore ist nicht endokrin aktiv (nichtfunktionelle Tumore).
Wachstum
Bei neuroendokrinen Tumoren handelt es sich meistens um hochdifferenzierte Tumore, die nur langsam wachsen und bei ihrer Erkennung oft noch gutartig sind. Sie können aber auch Zeichen einer Invasivität zeigen und werden dann als neuroendokrine Karzinome bezeichnet.
Gering differenzierte, neuroendokrine Karzinome dagegen können früh Metastasen bilden. Solange sie im Einzugsbereich der Pfortader liegen, sind sie symptomarm oder symptomlos, da die Leber die von ihnen gebildeten Mediatorstoffe entsorgt. Wenn sie in die Leber metastasieren, beginnt eine mehr oder weniger ausgeprägte Symptomatik, die von der Art ihrer Hormon- und Mediatorproduktion abhängt.
Diagnostik
Häufig werden neuroendokrine Tumore erst relativ spät erkannt; sie machen sich durch ihre Größe oder durch hormonelle Effekte, die jedoch nicht immer vorhanden sind, bemerkbar.
Bildgebende Verfahren
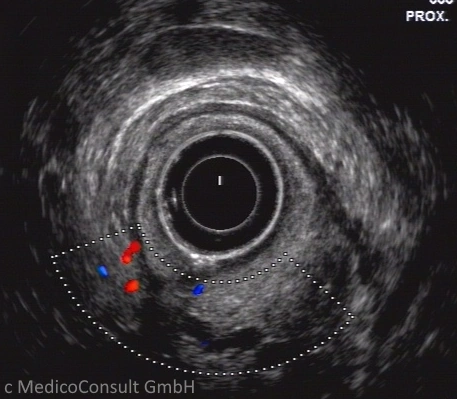
Die Abklärung erfolgt durch bildgebende Verfahren wie Sonographie, Computertomographie, MRT oder PET. Ist die Diagnose histologisch (nach Probepunktion) gestellt, schließt sich eine Ausbreitungsdiagnostik (meistens durch eine Somatostatin-Rezeptor-Sinitigraphie) an.
Marker
Chromogranin A ist ein sensitiver Marker für alle neuroendokrinen Tumoren; sie sind insbesondere bei Metastasierung erhöht.
5-Hydroxyindolessigsäure (5-HIES): findet sich beim Karzinoid-Syndrom im Urin erhöht.
Gastrin
Gastrin: Gastrinwerte von >1000 pg/ml weisen auf ein Gastrinom hin.
Insulin
Insulin: Im Fastentest zeigt sich ein deutlich erhöhter basaler Insulinspiegel (in µM/ml) im Verhältnis zur Glukose i.S. (in mg/dl) (Quotient > 0,3).
Somatostatinrezeptor-Szintigraphie:
Sie ist in vielen Fällen positiv bei Karzinoidtumoren und endokrinen Pankreastumoren und kann auch Metastasen nachweisen (Tumorgröße ab etwa 1 cm).
Bildgebende Verfahren
Sonographie, MRT und CT sind oft bei der Diagnostik des Primärtumors nicht erfolgreich, da er sehr klein sein kann. Eine Endosonographie kann dagegen erfolgreich sein. In jedem Fall wird man die bildgebenden Möglichkeiten bei Verdacht auch einen neuroendokrinen Tumor ausschöpfen.
Klassifikation
Neuroendokrine Tumore lassen sich histologisch folgendermaßen klassifizieren:
1a: Hoch differenzierter neuroendokriner Tumor
1b: Hoch differenziertes neuroendokrines Karzinom
2: Niedrig differenziertes neuroendokrines Karzinom
Für neuroendokrine Malignome wird ein differenziertes Stagingsystem vergewendet, das für chirurgische Therapieoptionen verwendet wird 1.
Einteilung
Klinische Bedeutung haben folgende Unterscheidungen:
| Tumor | Symptomatik | Lokalisation |
| Karzinoid | Flush, Diarrhö, Hypotonie | Gastrointestinaltrakt, Lunge |
| Gastrinom (Zollinger-Ellison Syndrom) | Peptische Geschwüre, Diarrhö | Pankreas, Duodenum, Magen |
| Insulinom | Hypoglykämie | Pankreas |
| Glukagonom | Diabetes mellitus, wanderndes nekrolytisches Erythem | Pankreas |
| VIPom (Verner-Morrison-Syndrom) | Wässrige Diarrhö, Hypokaliämie, Hyperkalzämie | Pankreas |
| Somatostatinom | Diabetes mellitus, Steatorrhö, Cholelithiasis | Pankreas, Dünndarm |
| Neuroblastom | Neurologische Symptomatik | ZNS |
| Medulläres Schilddrüsenkarzinom | Hypokalzämie | Schilddrüse |
| Melanom | lokale Symptomatik | Haut |
| Phäochromozytom | Blutdruckkrisen | Nebennierenmark |
Die ersten 6 neuroendokrinen Tumore der Tabelle sind in der Regel mit dem Intestinaltrakt assoziiert und werden als „gastroenteropankreatische neuroendokrine Tumore“ (GET-NET) zusammengefasst. Sie finden sich besonders in Magen, Duodenum und Appendix, seltener im Pankreas und sehr selten im Kolon oder Rektum.
Nach der WHO-Klassifikation aus dem Jahr 2000 ist der Begriff „Karzinoid“ zugunsten der Begriffe „neuroendokriner Tumor“ oder „neuroendokrines Karzinom“ fallen gelassen worden 2. Karzinoid-Tumoren des mittleren Darmbereichs (Jejunum, Ileum, Blinddarm) verursachen die Symptomatik des Karzinoid-Syndroms, die des unteren Darmabschnitts (Dickdarm) bleiben lange symptomlos.
Dignität
NET des Magens
Vorkommen von neuroendokrinen Tumoren (NET) des Magens oft zusammen mit einer Typ-A-Gastritis (Folge: Achlorhydrie, dadurch ständige G-Zell-Stimulation, daraufhin durch Hypergastrinämie Förderung des Wachstums der endokrin aktiven ECL-Zellen der Korpusschleimhaut mit Hyperplasieherden, aus denen später Tumore hervorgehen können). Sie sind bei ihrer Entdeckung durch Gastroskopie meist noch klein und gutartig und können vollständig abgetragen werden. Bei einigen Fällen lässt sich eine multiple endokrine Neoplasie (MEN Typ 1) diagnostizieren. Tumore mit einem Durchmesser vom mehr als 2 cm sind verdächtig auf Infiltration der Muscularis propria und Lymphknotenmetastasen.
NET der oberen Dünndarms
In 2/3 der Fälle Gastrinome mit ZES-Symptomatik, seltener (ca. 15 %) Somatostatinome und sonstige NET. Gastrinome, die mit MEN-Typ 1 assoziiert sind, treten oft multipel auf, sporadische meist einzeln. Duodenale Gastrinome metastasieren früh in regionale Lymphknoten und das umgebende Pankreas. Bei den seltenen pankreatischen Gastrinomen finden sich frühzeitig Lebermetastasen.
Somatostatinome liegen häufig im Bereich der Papilla Vateri. Sie sind häufig assoziiert mit einer Neurofibromatose (Typ 1) und selbst – im Gegensatz zu den pankreatischen Somatostatinomen – nicht endokrin aktiv.
NET des Ileums
Sie werden oft erst ab einer Größe von über 2,5 cm klinisch auffällig und weisen dann oft schon Lymphknoten- und Lebermetastasen mit einem Karzinoid-Syndrom (Flush, Diarrhö, Endokardfibrose) auf.
NET der Appendix
Sie sind häufig benigne; ab einer Größe von > 2,5 cm muss mit Malignität gerechnet werden.
NET des Kolons
Sie sind selten und haben bei Diagnosestellung häufig bereits Metastasen. NET im Rektum sind, da sie meistens hochdifferenziert sind, prognostisch relativ günstig.
NET des Pankreas
Bei der Diagnosestellung sind die Tumore häufig bereits zu Karzinomen entartet (Ausnahme: Insulinom, das bis 2 cm Durchmesser meistens gutartig ist). Etwa die Hälfte von ihnen ist endokrin aktiv (Zollinger-Ellison-Syndrom, VIPom-Syndrom, Insulinom-Syndrom, Glukagonom-Syndrom, Verner-Morrison-Syndrom). Häufig produzieren die Tumorzellen mehr als ein hormonelles Prinzip; benannt werden sie nach der klinisch vorherrschenden Symptomatik. Malignitätskriterien sind eine Größe von > 2 cm, Wachstum und nachweisbare Metastasen. Bei Nachweis von Gefäßeinsprossungen muss von Malignität ausgegangen werden. Glukagonome sind bei einer Größe von > 2 cm in 60 % metastasiert.
Therapie
- Eine frühzeitige Operation kann zu einer Heilung führen. Operationen sind ansonsten in Einzelfällen zur Tumorverkleinerung und zur Komplikationsbeherrschung indiziert.
- Lokal ablative Verfahren (z. B. Elektrokoagulation oder Radioembolisation von Lebermetastasen, SIRT).
- Analoga von Somatostatin vermindern vielfach die hormonell bedingten Beschwerden deutlich.
- Eine Chemotherapie wird bei rasch wachsenden, wenig differenzierten Tumoren erforderlich.
- Carbozatinib ist ein Tyrosin-Kinase-Inhibitor (TKI), der bei neuroendokrinen Tumoren des Bauchraums in einer Studie das progressionfreie Überleben auf 8,4 Monate (vs. 3,9 Monate unter Placebo) erhöht hat 3.
- Die Peptid-Receptor-Radiotherapie ist ein Verfahren, bei dem radioaktive Liganden an tumorspezifische Rezeptoren binden und eine lokale Bestrahlung verursachen. 4 Als erfolgreich wird die Therapie mit 177Lu-DOTATATE (177Lutetium-[DOTA°,Tyr3]octreotate) beschrieben. 5
→ Siehe unter APUDome.
Hereditäre neuroendokrine Tumore
Das gemeinsame Auftreten neuroendokriner Tumore in verschiedenen Organen wird unter Multiple endokrine Neoplasie zusammengefasst. Sie weist in der Regel eine familiäre Häufung und genetische Ursache auf. Es werden zwei Typen unterschieden:
- Multiple endokrine Neoplasie Typ 1 (MEN Typ 1): neuroendokrine Tumore gehäuft in Hypophysenvorderlappen (meist gutartige Adenome), Nebenschilddrüse, Pankreas, Duodenum, z. B. Insulinom, Glukagonom, Gastrinom, Karzinoid, VIPom.
- Multiple endokrine Neoplasie Typ 2 (MEN Typ 2) (seltener als Typ 1): praktisch immer ein medulläres Schilddrüsenkarzinom, in etwa 50 % ein Phäochromozytom, in etwa 20 % ein primärer Hyperparathyreoidismus bei Nebenschilddrüsenadenom.
→ Zur multiplen endokrinen Neoplasie siehe hier.
Verweise
- APUDome
- Multiple endokrine Neoplasie
- Karzinoid
- Gastrinom
- Insulinom
- Gastrinom
- Phäochromozytom
- VIPom
- Medulläres Schilddrüsenkarzinom
- Primärer Hyperparathyreoidismus
- Tumorsuche
- Das Hormonsystem: Basics
Weiteres
- Pancreas 2019;48:613–21[↩]
- Ann N Y Acad Sci. 2004 Apr;1014:13-27. doi: 10.1196/annals.1294.002.[↩]
- N Engl J Med. 2025 Feb 13;392(7):653-665. doi: 10.1056/NEJMoa2403991[↩]
- Drugs Today (Barc). 2011 Oct;47(10):773-86[↩]
- Expert Rev Gastroenterol Hepatol. 2019 Nov;13(11):1023-1031. DOI: 10.1080/17474124.2019.1685381[↩]