Allgemeines
Das Long-QT-Syndrom inkl. Romano-Ward- und das Jerwell-Lange-Nielsen-Syndrom sind angeborene Veranlagungen für bösartige und potenziell tödliche Herzrhythmusstörungen auf dem Boden einer Verlängerung der QT-Zeit.
Das angeborene Long-QT-Syndrom (LQTS) beruht auf einer angeborenen Veränderung des Kalziumkanals in der Zellmembran der Herzmuskelzellen, die zu einem verlängerten QT-Intervall und einer verzögerten Repolarisation führt. Die Prävalenz liegt bei etwa 1:2000. 1
Ist die QT-Verlängerung zudem mit angeborener Schwerhörigkeit oder Taubheit verbunden, entspricht dies dem Jervell-Lange-Nielsen-Syndrom, das autosomal rezessiv vererbt wird und mit einem besonders hohen Risiko für maligne Herzrhythmusstörungen verbunden ist. 2 3
Die durch die Kalziumkanal-Anomalie hervorgerufenen Herzrhythmusstörungen können in Form von Torsade-de pointes-Tachykardien und Kammerflimmern lebensbedrohlich sein.
Körperliche Aktivitäten und psychischer Stress sowie Antipsychotika können das Risiko von tödlichem Kammerflimmern erheblich erhöhen. 4
Die Lebenserwartung beim Long-QT-Syndrom ist erheblich eingeschränkt, ist aber abhängig vom genetischen Typ. Die Mutation KCNQ1-A341V hat die schlechteste Prognose von durchschnittlich kaum 10 Jahren. 3
Genetik
Das Long-QT-Syndrom Typ 1 (LQT1) ist der häufigste Typ der Long-QT-Syndrome (LQTS). Es beruht auf Mutationen der Gene KCNQ1, KCNH2, KCNE1, KCNE2, CACNA1c, CAV3, SCN5A, SCN4B. Die häufigsten LQTS-Gene sind bei weitem KCNQ1 (LQT1), KCNH2 (LQT2) und SCN5A (LQT3). 5 3
Mutationen beider Allele ohne Taubheit werden als Romano-Ward-Syndrom (AR RWS) bezeichnet, mit Taubheit dagegen als Jervell- und Lange-Nielsen-Syndrom (JLNS). 6
Inzwischen wurden bei Patienten über 700 KCNQ1-Varianten identifiziert. Die überwiegende Mehrheit wird dominant vererbt. 7
LQT1 wird autosomal-dominant vererbt, wohingegen JLNS1 autosomal-rezessiv vererbt wird und sporadisch eine zusammengesetzte Heterozygotie aufweist. 8 9
Das menschliche KCNQ1-Gen (Kaliumkanal, spannungsgesteuert, KQT-ähnliche Unterfamilie, Mitglied 1) weist zwei Transkript-Isoformen auf, eine Nieren- und eine Pankreas-Isoform. Beide können einen funktionellen kardialen Kaliumkanal bilden und werden im menschlichen Herzen in erheblichen Mengen exprimiert. 10
Diagnostik
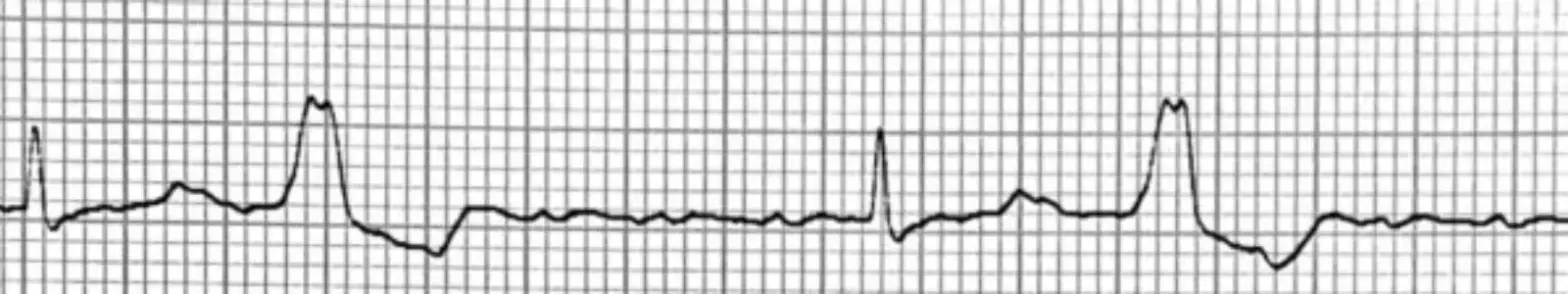
Die Diagnose beruht auf einer Verlängerung der QT-Zeit über ≥480 msek im Elektrokardiogramm (EKG). Für die Diagnostik spielt die QT-Verlängerung in der Erholungsphase eines Belastungstests eine besondere Rolle. Eine Bestätigung erfolgt durch einen Gentest. 3
Therapie
Medikamente: Die Behandlung beruht auf einer Unterdrückung des Einflusses von Stress auf das Herz durch ß-Blocker (β-adrenerge Blockade). Bevorzugt werden lang wirkende Substanzen. Nadolol ermöglicht eine zweimal tägliche Verabreichung, normalerweise mit 1 bis 1,5 mg/kg pro Tag. Metoprolol wird als weniger wirksam nicht empfohlen. Betablocker sind bei LQT1-Patienten ganz besonders wirksam. Bei LQT2-Patienten trotz β-Blockern werden mehr lebensbedrohliche Ereignisse (vor allem Herzstillstände) beschrieben. 3
Eine sympathische Denervierung des linken Herzens ist eine zunehmend genutzte Alternative zur Prophylaxe lebensbedrohlicher Rhythmusstörungen. Der Eingriff kann video-thorakoskopisch erfolgen. 11 12 13
→ Auf facebook informieren wir Sie über Neues und Interessantes.
→ Verwalten Sie Ihre Laborwerte mit der Labor-App Blutwerte PRO – mit Lexikonfunktion.
Verweise
Weiteres
- Circulation. 2009;120:1761–1767[↩]
- Am. Heart J. 1957;54:59–68[↩]
- Circ Arrhythm Electrophysiol. 2012 Dec;5(6):e119-20[↩][↩][↩][↩][↩]
- Am J Health Syst Pharm. 2008 Jun 1;65(11):1029-38[↩]
- J Rare Dis. 2008 Jul 7;3:18. doi: 10.1186/1750-1172-3-18[↩]
- Am J Med Genet A. 2016 Jun;170(6):1510-9. DOI: 10.1002/ajmg.a.37636[↩]
- Biomedicines. 2022 Sep 12;10(9):2254. doi: 10.3390/biomedicines10092254[↩]
- Circ Arrhythm Electrophysiol. 2012;5(4):868–77. doi: 10.1161/CIRCEP.111.962019[↩]
- Int J Mol Sci. 2023 Jan 10;24(2):1350. doi: 10.3390/ijms24021350[↩]
- BMC Med Genet. 2017 Jun 8;18(1):66. DOI: 10.1186/s12881-017-0430-7[↩]
- Thorac Dis. 2017 Sep;9(9):3394-3397[↩]
- Asian Cardiovasc Thorac Ann. 2021 Mar;29(3):186-190.[↩]
- JACC Clin Electrophysiol. 2022 Mar;8(3):281-294[↩]