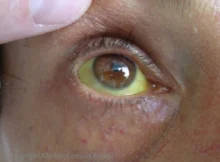
Bilirubin ist ein gelber Blutfarbstoff, der beim Abbau roter Blutkörperchen aus dem roten Blutfarbstoffs Hämoglobin hervorgeht. Es wird durch die Leber in die Galle ausgeschieden und ist als „Gallefarbstoff“ für deren grünlich-gelbe Farbe verantwortlich. Bei Leberkrankheiten kann die Bilirubinausscheidung so beeinträchtigt werden, dass es zu einer Erhöhung der Blutkonzentration und damit zu einer Gelbsucht (Ikterus) kommt. Diagnostisch wird Bilirubin zur Beurteilung eines Blutzerfalls (Hämolyse) oder eines Gallestaus (Cholestase) verwendet.
Unterscheidung von direktem und indirektem Bilirubin: Direktes Bilirubin ist durch die Leber wasserlöslich gemacht worden ist; indirektes ist durch einen vermehrten Blutzerfall entstanden und wird im Blut an Eiweiß gebunden transportiert.
→ Hämoglobin
→ Gelbsucht (Ikterus)
→ Leberkrankheiten – einfach erklärt
Normwerte
- Gesamtbilirubin im Blut: oberer Grenzwert 1,1 mg/dl (< 19 µmol/l)
- Direktes Bilirubin (konjugiertes, wasserlösliches Bilirubin): oberer Grenzwert 0,25 mg/dl, andere Labore 0,6 mg/dl (< 10 µmol/l)
- Indirektes Bilirubin: kann aus Differenz zwischen Gesamtbilirubin und direktem Bilirubin abgeschätzt werden.
- Neugeborene: Anstieg des Gesamtbilirubins bis 13,5 mg/dl am 3. – 5. Tag, danach Abfall. Werte darüber bedürfen intensiver Beobachtung.
Interpretation der Bilirubinwerte
- Ein Anstieg des Gesamtbilirubins mit überwiegendem Anteil an unkonjugiertem Bilirubin ist ein Hinweis auf einen vermehrten Blutzerfall (Hämolyse). Sind die Transaminasen und alkalische Phosphatase normal, dann kann es sich um eine funktionelle Hyperbilirubinämie (z. B. Meulengracht-Syndrom, Criggler-Najjar-Syndrom) handeln, oder um eine Hämolyse (dabei Haptoglobin erniedrigt und LDH stark erhöht).
- Eine Erhöhung des Geamtbilirubins ohne deutliche Erhöhung auch des indirekten Bilirubins weist auf eine Leberkrankheit mit Cholestase hin. Sie ist abzusichern durch Bestimmung der Leberwerte, insbesondere der Cholestaseenzyme. Zu Lebererkrankungen siehe hier.
Struktur, Stoffwechsel
Bilirubin ist ein extrem wasserunlösliches Tetrapyrrol, das aus dem Hämabbau hervorgeht. Seine Hauptquelle ist der Hämoglobinabbau aus alternden Erythrozyten. Bildungsorte sind vor allem die Zellen des retikuloendothelialen Systems (RES). Von dort wird es an Albumin gebunden zur Leber transportiert, in der es aus dem Blut extrahiert und biliär (in die Galle) sezerniert wird. Die hepatobiliäre Sekretion umfasst schematisch:
- Transportvorgänge an der sinulateralen Zellmembran der Leberzellen,
- intrazelluläre Bindung und Stoffwechsel,
- die Ausscheidung aus den Leberzellen (Hepatozyten) in die Gallenkapillaren.
Aufnahme in die Leberzellen
Die Aufnahme von Bilirubin in die Hepatozyten erfolgt Carrier-vermittelt, energie- und chloridabhängig. Sie ist sehr rasch und stellt im Gesamttransport aus dem Blut in die Galle nicht den geschwindigkeitslimitierenden Schritt dar. Aus kinetischen Untersuchungen wird auf einen erheblichen Reflux (etwa 40 %) aus der Leberzelle (Hepatozyt) in das Blut geschlossen, der in Fällen unkonjugierter Hyperbilirubinämie noch weiter gesteigert sein kann. Bromsulphthalein (BSP) und Indozyaningrün (ICG), werden über das gleiche Transportsystem aufgenommen, Gallensäuren dagegen nicht. Gallensäuren werden nur bei einer Cholestase, nicht dagegen bei den funktionellen Hyperbilirubinämiesyndromen im Blut retiniert. Mit ihrer Hilfe können cholestatisch und nicht-cholestatisch bedingte Hyperbilirubinämien differenziert werden.
Intrazellulärer Stoffwechsel
Intrazellulär wird Bilirubin an Ligandin und das Z-Protein gebunden. Die Konzentration dieser Proteine bestimmt die Bindungskapazität der Leber für Bilirubin. Auf ungeklärte Weise gelangt Bilirubin ins endoplasmatische Retikulum, wo es durch eine Bilirubin-UDP-Glukuronyltransferase konjugiert wird. Das Enzym besteht aus 4 Untereinheiten, die alle 4 zur Bildung von Diglucuroniden benötigt werden. Zur Bildung von Monoglukuroniden reicht dagegen eine Untereinheit aus. Man nimmt an, dass ein geringer Anteil der Glukuronide durch eine ß-Glukuronidase oder in einer Rückreaktion der Transferasereaktion wieder dekonjugiert wird, sodass Monoglukuronide und unkonjugiertes Bilirubin entstehen können. Welche Bedeutung diese Reaktionen für die Ausbildung unkonjugierter Hyperbilirubinämien besitzen, ist unbekannt.
Biliäre Ausscheidung
Die Ausscheidung in die Gallenkapillaren schließlich geschieht carriervermittelt gegen einen Konzentrationsgradienten und bedarf der Energiezufuhr. Das gallenkapilläre Transportsystem scheint für Bilirubin nicht spezifisch zu sein. Es ist auch zuständig für die biliäre Sekretion anderer organischer Anionen wie BSP, ICG, Bengalrosa, Methylenblau, nicht dagegen für Gallensäuren. Es transportiert auch unkonjugiertes Bilirubin. So findet sich in der Galle praktisch ausschließlich konjugiertes Bilirubin, zu 80 % als Di- und zu 20 % als Monoglukuronid.
Störungen von Transport und Stoffwechsel
Der Weg des Bilirubins durch die Leberzelle kann auf der Ebene eines jeden Teilschritts gestört sein:
- Defekt bei der Aufnahme des Bilirubins aus dem Blut in die Leberzelle.
- Die Aufnahmegeschwindigkeit von Bilirubin in die Leberzelle ist beim Gilbert-Meulengracht-Syndrom– und möglicherweise auch beim Rotor-Syndrom erniedrigt.
- Defekt bei der intrazellulären Speicherung
- Eine Erniedrigung der intrazellulären Speicherkapazität für Bilirubin und auch für BSP findet sich beim Rotor-Syndrom.
- Defekt bei der Glukuronidierung des Bilirubins.
- Beim Crigler-Najjar-Syndrom Typ I fehlt die Aktivität der Bilirubin-UDP-Glucuronyltransferase vollständig.
- Beim Crigler-Najjar-Syndrom Typ II ist sie auf etwa 10 % und beim Gilbert-Syndrom auf etwa 50 % herabgesetzt.
- Defekt bei der biliären Sekretion.
- Diese Störung liegt dem Dubin-Johnson-Syndrom zugrunde.
Beim Ikterus im Rahmen einer Leberentzündung (Hepatitis) oder einer toxischen Leberschädigung liegt eine Funktionsstörung der Leberzellen (hepatozelluläre Cholestase) vor, bei der jedoch die Konjugationsschritte noch weitgehend intakt sind. Das Serumbilirubin ist direkt reagierend (glukuronidiert). Beim zunehmenden Leberversagen kann das indirekt reagierende Bilirubin (nicht glukuronidiert) zu überwiegen beginnen.
→ facebook: Neues und Interessantes!
→ Labor-App Blutwerte PRO – mit Lexikonfunktion
Verweise
Dieser Beitrag ist Teil unseres Themenbereichs Leberzirrhose.
- Laborwerte bei Leberkrankheiten
- Cholestaseparameter
- Cholestase
- Hämolyse
- Gilbert-Meulengracht-Syndrom
- Galle
Patienteninfos
Weiteres