Die kongenitale erythropoetische Porphyrie (CEP, Morbus Günther) ist eine sehr seltene genetisch determinierte Erkrankung des Porphyrinstoffwechsels, die autosomal-rezessiv vererbt wird und zu ausgeprägten bullös-entzündlichen Hautveränderungen an lichtexponierten Stellen (Photodermatose) führt. Sie ist bei Namensähnlichkeit von der häufigeren „erythropoetischen Protoporphyrie“ (EPP) zu unterscheiden, einer Porphyrie durch Defekt der mitochondrialen Hämsynthase bzw. Ferrochelatase (FECH), bei der im Unterschied zur erythropoetischen Porphyrie die Zähne nicht fluoreszieren, kaum narbige Hautläsionen entstehen und sich eine Leberzirrhose entwickelt. Die Behandlung besteht im Wesentlichen in einer strikten Vermeidung einer Lichtexposition (Sonnenschutzkleidung, Sonnenbrille sowie gefärbten Auto- und Fensterscheiben). 1
Entstehung
Die Erkrankung ist gegenisch determiniert und wird autosomal rezessiv weitergegeben. Die genetische Grundlage der erythropoetischen Porphyrie ist eine Mutation des Enzyms URO-III-Synthase (UROIIIS); inzwischen sind mehr als 25 Mutationen bekannt, die häufigste in etwa 1/3 der Fälle ist C73R (Cystein ersetzt durch Arginin). Die Mutanten weisen je nach Mutation eine unterschiedliche enzymatische Restaktivität auf 2, so dass noch geringe Mengen von UROIII gebildet werden können. Entsprechend sind die klinischen Manifestationen in ihrem Schweregrad unterschiedlich. Die Mutanten sind jedoch instabil, verändern ihre Faltung und werden abgebaut.
Im Fall der C73R-Mutation leidet ebenfalls nicht die enzymatische Aktivität sondern die Stabilität des Enzyms. Das falsch gefaltete Protein setzt einen Degradationsmechanismus über eine Proteasom-Induktion in Gang, so dass die Konzentration des Enzyms unter die Nachweisgrenze sinkt. Eine Behandlung mit dem Proteasominhibitor MG132 führt zu einer Anhebung des Enzymspiegels und einer Wiederherstellung der enzymatischen Aktivität in Zellkulturen. 3
Das sich bei UROIIIS-Mutation anhäufende Ausgangssubstrat Tetrapyrrolhydroxymethylbilan degradiert spontan vermehrt zu Uroporphyrin I, das im Körper akkumuliert, besonders in Knochen, Zähnen, Haut und Erythrozyten. Große Mengen an Porphyrinen werden über den Urin und den Stuhl ausgeschieden.
Symptomatik
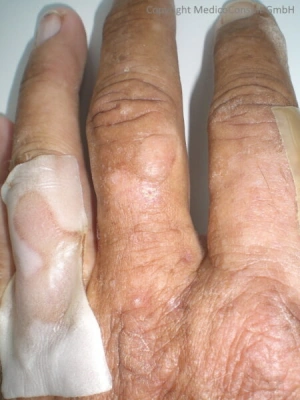
In schweren Fällen findet sich bereits pränatal oder perinatal eine ausgeprägte hämolytische Anämie mit sich entwickelnder pulmonaler Hypertonie; auch wird ein Hydrops fetalis beschrieben 4 5. Leichtere Fälle können sich auf die Hautmanifestationen beschränken. An der Haut kommt es durch Lichteinwirkung zu oxidativen Prozessen mit starken Schädigungen, bullösen Hautdefekten und anschließenden narbigen Abheilungen. Typischerweise sind Hände, Gesicht und Ausschnitt sind mit kleinen weißlichen Narben übersät. Oft findet sich ein Hirsutismus (vermehrte Körperbehaarung) oder eine faciale Hypertrichose (vermehrte Behaarung im Gesicht).
Diagnostik
Bei einer erythropoetischen Porphyrie fällt gelegentlich eine rötliche Verfärbung der Zähne auf (Erythrodontie). Unter der uv-Lampe fluoreszieren Zähne, Urin und Blut kräftig rot. Im Urin werden große Mengen an Porphyrinen gemessen (lichtundurchlässiges Sammelgefäß).
Die Diagnose wird durch genetische Analyse bestätigt.
Therapie
Die erythropoetische Porphyrie ist schwer behandelbar. Licht wird vermieden, und die Betroffenen werden nachtaktiv. Eine Basispflege der Haut zur Vermeidung von Superinfektionen und Heilung offener Läsionen ist erforderlich. Der bei Lichtmangel eintretende Vitamin-D-Mangel muss ausgeglichen werden.
Eine Heilung der erythropoetischen Porphyrie lässt sich nur durch Übertragung des gesunden UROIIIS-Gens erreichen, was prinzipiell über eine Knochenmarktransplantation oder über einen Vektor (einen retroviral-vermittelten Gentransfer) erfolgen kann.
- Knochenmarktransplantation: Die allogene hämatopoetische Stammzelltransplantation führt zur Heilung der erythropoetischen Porphyrie 6, ist jedoch mit Risiken verbunden, so kann z. B. eine kutane Graft-versus-Host-Reaktion eintreten 7, was bei der Indikationsstellung zu berücksichtigen ist.
- Behandlung mit einem Gen-Vektor: Die Möglichkeit einer retroviralen Gen-Therapie wurde durch erfolgreiche Untersuchungen an Kulturen transduzierter Zelllinien inauguriert 8 2. Allerdings besteht die Gefahr der Entstehung einer Leukämie, was einer Übertragung dieser Behandlungsmethode auf den Menschen im Wege steht.
- Behandlung von aus Keratinozyten induzierten pluripotenten Stammzellen des Patienten, bei denen eine Genkorrektur durch einen Lentivirus-Vektor, der UROS-cDNA enthielt, vorgenommen wurde. Erythroblasten, die auf diese Weise behandelt wurden, verloren als Zeichen ihrer Heilung ihre Porphyrie-bedingte Fluoreszenz 9. Dies eröffnet eine viel versprechende neue Therapiemöglichkeit.
Eine Verbesserung der Symptomatik ist möglicherweise in vielen Fällen durch Hemmung des Abbaus des defekten Enzyms UROIIIS möglich, wie in-vitro-Untersuchungen hoffen lassen (s.o.). 10
→ Auf facebook informieren wir Sie über Neues und Interessantes!
→ Verwalten Sie Ihre Laborwerte mit der Labor-App Blutwerte PRO – mit Lexikonfunktion.
Verweise
- Mol Genet Metab. 2019 Nov;128(3):288-297. DOI: 10.1016/j.ymgme.2018.12.008 . Epub 2018 Dec 27. PMID: 30685241; PMCID: PMC6597325.[↩]
- Biochemistry. 2009 Jan 20;48(2):454-61[↩][↩]
- J Biol Chem. 2011 April 15; 286(15): 13127–13133[↩]
- Prenat Diagn. 2003 Jan;23(1):25-30[↩]
- Prenat Diagn. 2004 Apr;24(4):282-6[↩]
- Ann Dermatol Venereol 2010;137:635-9v[↩]
- Br J Dermatol. 2007 Mar;156(3):567-71[↩]
- Mol Genet Metab. 1998 Sep;65(1):10-7[↩]
- Am J Hum Genet. 2012 Jul 13;91(1):109-21[↩]
- J Biol Chem. 2011 Apr 15;286(15):13127-33[↩]