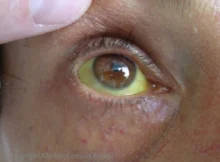
Unter dem Begriff „familiäre intrahepatische Cholestase“ (progressive familial intrahepatic cholestasis (PFIC)) sind verschiedene autosomal rezessiv vererbte, fortschreitende Erkrankung der Leber zusammengefasst, bei der die Bildung der Galle gestört ist. Folgen sind ein Aufstau von Gallensäuren und anderer gallepflichtiger Substanzen im Körper und die Entwicklung einer Leberzirrhose schon im Kindesalter.
Verschiedene Typen
Familiäre intrahepatische Cholestase ist eine heterogene Erkrankungsgruppe. Inzwischen sind drei Gendefekte als Ursache festgestellt worden 2:
- PFIC1: Byler-Syndrom, Gen ATP8B1: Mutation führt indirekt zu Störung der Gallebildung.
- PFIC2: BSEP-Defizienz, Gen ABCB11: Mutation führt zu mangelhafter Gallensäureausscheidung in die Galle.
- PFIC3: MDR3-Defizienz, Gen ABCB4, Mutation führt zu mangelhaftem Phospholipid-Transport in die Galle.
Epidemiologie, Häufigkeit
Etwa 10 – 15 % der kindlichen Cholestasen sollen auf eine familiäre intrahepatische Cholestase zurückzuführen sein. wovon 2/3 auf PFIC1 und PFIC2 und 1/3 auf PFIC3 fallen sollen. Vermutlich liegt die Inzidenz der familiären intrahepatischen Cholestasen bei 1: 50-100.000 Geburten; beide Geschlechter sind etwa gleich häufig betroffen.
Stoffwechseldefekt und Auswirkungen
Alle Formen der intrahepatischen Cholestase führen zu einer schweren Ausscheidungsstörung von Gallensäuren und anderen gallepflichtigen Verbindungen. Diese Verbindungen sammeln sich in den Hepatozyten an und stauen sich weiter zurück in den Körper.
- PFIC1: Das ATP8B1-Gen ATP (Chromosom 18) codiert für einen Aminophospholipid-Transporter und versorgt die kanalikuläre Seite der Plasmamembran der Hepatozyten mit Phosphatidylserin und Phosphatidyläthanolamin. Dies trägt zum Schutz der Membran vor hoher Gallensäurekonzentration bei. Die Mutation führt damit zu einem mangelhaften Schutz, was zur Cholestase führt. Das ATP8B1-Gen findet sich nicht nur in der Leber, sondern auch in der Bauchspeicheldrüse, den Nieren und dem Dünndarm, was die extrahepatische Symptomatik (s. u.) erklärt.
- PFIC2: Das ABCB11-Gen (Chromosom 2) codiert für eine ATP-abhängige Gallensäure-Exportpumpe (BSEP) an der Gallenkapillarmembran der Hepatozyten. Es gibt offenbar verschiedene Mutationen, die alle zu einer Funktionslosigkeit des Gallensäuretransports führen.
- PFIC3: Die Mutation im ABCB4-Gen (Chromosom 7) bewirkt eine reduzierte Ausscheidung von Phosphatidylcholin (Lecithin) in die Galle. Lecithin bildet mit Gallensäuren gemischte Mizellen. Es senkt auf diese Weise deren freie Konzentration, was die Epithelzellen der Gallenwege vor ihrer toxischen Wirkung schützt. Wenn die Konzentration freier Gallensäuren in der Galle bei defektem Lecithintransport in die Galle steigt, so führt dies zu einer chronischen Entzündung (chronische Cholangitis), was schließlich zur Leberzirrhose führt. Da Lecithin zur Lösung von Cholesterin erforderlich ist, bedeutet eine reduzierte Lecithin-Ausscheidung auch gleichzeitig eine Reduktion der Löslichkeit von Cholesterin (siehe hier). Die Galle wird an Cholesterin übersättigt; es kommt zur Bildung von Cholesterinkristallen und schließlich zur Bildung von Gallensteinen.
Klinische Symptome
Nach einer Zusammenstellung berichteter Fälle 2 sollen sich die verschiedenen Formen der familiären intrahepatischen Cholestase klinisch leicht voneinander unterscheiden:
- PFIC1 Byler-Syndrom: schwerer Juckreiz; in den ersten Lebensmonaten erst episodische, dann dauerhafte Gelbsucht, Gedeihstörung mit starker Wachstumsverzögerung, wässrige Diarrhö, Pankreatitis, Schwerhörigkeit.
- PFIC2: schwerer Juckreiz; gleich dauerhafte Gelbsucht, Gallensteine, Neigung zu frühem hepatozellulärem Karzinom (oft schon im ersten Lebensjahr).
- PFIC3: nur milder bis mäßiger Juckreiz. Langsamerer Verlauf als bei PFIC1 und PFIC2. Cholestasezeichen noch kaum perinatal vorhanden, im ersten Lebensjahr in etwa 1/3 der Fälle. Später sekundäre Symptome einer Leberzirrhose mit portaler Hypertension. Ein Lebertumor (wie beim PFIC2) kommt nicht oder kaum vor.
Diagnostik
Eine unerklärte Cholestase bei Säuglingen und Kinder (ohne Gallengangsobstruktion oder Hepatitis) sollte an eine seltene familiäre intrahepatische Cholestase denken lassen.
Sonographie
Die Gallenwege sind sonographisch nicht gestaut.
Laborwerte
Die Gamma-GT-Werte im Serum sind bei PFIC1 und 2 normal, bei PFIC3 stark erhöht. Die Gallensäurekonzentrationen im Blut sind bei PFIC1 und 2 sehr hoch, bei PFIC3 erreichen sie nicht ganz so hohe Werte.
Histologie
Auffällig ist, dass PFIC1 und 2 keine Gallengangsproliferationen entwickeln, während PFIC3 ausgeprägte duktuläre Proliferationen in den Portalfeldern aufweisen. Der Fibrosegrad ist je nach Stadium unterschiedlich.
Genetische Analyse
Eine genetische Analyse bei Kindern mit unerklärter chronischer intrahepatischer Cholestase führt zur Diagnose.
Therapie
Eine symptomatische Therapie aller Formen einer familiären intrahepatischen Cholestase, insbesondere von PFIC3, stellt UDCA (Ursodesoxycholsäure) dar. 3 Eine MCT-Kost (mittelkettige Triglyceride) und die Zufuhr fettlöslicher Vitamine (A, D, E, K) gehören in das Therapiespektrum.
Die Therapie des Juckreizes stellt ein großes Problem dar. Gallepflichtige Medikamente können durch Retention zu erheblichen Nebenwirkungen führen.
Eine Lebertransplantation führt zur Heilung Transplantation. 1990 Nov;50(5):804-6, bei PFIC1 allerdings nicht zur Heilung der extrahepatischen Komplikationen wie einer Pankreatitis und der Diarrhö. Die 5-Jahres-Überlebensrate der Transplantate und der Transplantierten lag in einer kleinen Studie bei 83%. 4
→ Auf facebook informieren wir Sie über Neues und Interessantes!
→ Verwalten Sie Ihre Laborwerte mit der Labor-App Blutwerte PRO – mit Lexikonfunktion.