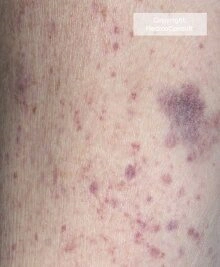
Immunthrombozytopenie (immunthrombozytopenische Purpura, früher: idiopathische thromboztopenische Purpura, ITP) bedeutet Mangel an Blutplättchen (Thrombozyten) durch Autoantikörper. Vom Immunsystem gebildete Antikörper greifen körpereigene Thrombozyten an. Der entstehende Thrombozytenmangel führt zu einer unzureichenden primären Blutstillung (Hämostase) mit der Folge spontan auftretender punktförmiger Einblutungen in Haut und Schleimhäute (Petechien).
Formen
Bezüglich der Genese werden eine primäre und eine sekundäre Form unterschieden. Die primäre Form ist definiert als eine isolierte Thrombozytopenie < 100 × 10ˆ9/l ohne erkennbare Ursachen. Die sekundäre Form lässt sich mit Auslösern, wie Infektionen, Medikamenten oder rheumatischen oder lymphoproliferativen Erkrankungen in Verbindung bringen.
Bezüglich des Verlaufs werden eine akute und eine chronische Form unterschieden. Die akute Form kommt häufiger bei Kindern vor; die chronische Form tritt eher bei Erwachsenen auf und wird hier auch als Morbus Werlhof bezeichnet 1.
Ursache

Die früher als idiopathisch bezeichnete thrombozytopenische Purpura ist heute meist nicht mehr „idiopathisch“, d. h. in ihrer Ursache unbekannt. Oft findet man eine Infektion mit Viren oder Helicobacter als Auslöser.
- Virusinfektionen als Auslöser 2: Beispiele sind Infektionen mit EBV und CMV. Man geht davon aus, dass es bei einer banalen Infektion zu einer Produktion von Antikörpern kommt, die eine Kreuzimmunität aufweisen. Zum einen richten sie sich gegen die körperfremden Pathogene, die eine Infektion im Körper auslösen und gleichzeitig aber auch gegen körpereigene Strukturen, nämlich die Thrombozyten. Dadurch werden sie zerstört und die absolute Anzahl der im Körper zirkulierenden Thrombozyten fällt stark ab (unter 100.000/µl, Blutungen ab ca. unter 30.000/µl, normal 150.000 bis 300.000/µl). Die Funktion in der primären Hämostase (Blutgerinnung) kann nicht mehr suffizient ausgeführt werden, und so kommt es zu kapillären Einblutungen in die Haut und Schleimhaut.
- Helicobacter pylori als Auslöser 3: Es besteht eine Assoziation der idiopathischen thrombozytopenischen Purpura (ITP) mit einer Infektion der Magenschleimhaut mit Helicobacter pylori, sodass bei der Diagosestellung einer ITP nach einer Helicobacter-Gastritis zu suchen ist. 4
Symptomatik
Ein banaler Infekt geht bei Kindern häufig einer ITP um ca. 3 bis 4 Wochen voraus. Ihr Allgemeinzustand ist in der Regel gut. Sie werden durch kleine und große Blutungen (Petechien und Hämatome) auffällig, die bei jeder kleinsten Kollision oder Verletzung entstehen. Auch finden sich im Mund eine schwärzlich belegte Zunge und Schleimhautblutungen, z. B. nach dem Zähneputzen.
Eine Blutungskomplikation unterschiedlicher Stärke kann zu akuten Gefährdungen führen. Bei älteren Patienten ≥ 70 Jahren können Thrombozytenwerte unter 20 – 30 × 10ˆ9/l zu einer erhöhten Gefahr auch von Hirnblutungen führen. Es wird daher geraten, das Alter bei einer Therapieentscheidung mit zu berücksichtigen. 1
Differenzialdiagnosen
Autoantikörper gegen Thrombozyten können auch im Rahmen der HIV-Infektion, einer Lymphomkrankheit oder des Lupus erythematodes auftreten.
Im Gegensatz zur Autoantikörper-induzierten idiopathischen thrombozytopenischen Purpura gibt es auch Thrombozytopenien, die primär durch eine Synthesestörung im Knochenmark hervorgerufen werden, z. B. durch Folsäuremangel, Vitamin-B12-Mangel oder im Rahmen einer Leukämie.
Eine Lymphomkrankheit, ein Lupus erythematodes, eine Leukämie und ein Mangel an Vitamin B12 oder Folsäure sind daher auszuschließen.
→ Morbus Werlhof
→ Thrombozytopenie
Diagnostik
Bei der Diagnostik einer ITP sind Blutwerte sowie eine Diagnostik auf EBV und CMV sowie auf eine Helicobacter-Gastritis einzubeziehen.
Unter den Laborwerten sind charakteristisch
- deutlich erniedrigte Blutplättchen (Thrombozyten), < 100 × 10ˆ9/l,
- normale plasmatische Gerinnungswerte,
- eine erhöhte Zahl an Megakaryozyten im Differenzialblutbild als Ausdruck der erhöhten Thrombozyten-Neubildung (Knochenmarkpunktion).
Therapie
Nicht jede ITP muss behandelt werden. Es wird eine individualisierte Vorgehensweise empfohlen. Zu berücksichtigen sind das Alter (s. o.), die individuelle Blutungsvorgeschichte, die Traumarisiken und ein eventuelles Risiko durch Therapeutika, wie ASS und NSAID. Ziel der Behandlung ist eine Reduktion des Blutungsrisikos durch Erhöhung der Thrombozytenzahl.
Vollständiges Ansprechen auf die Behandlung wird definiert als Thrombozytenzahl ≥ 100 x 10ˆ9/l und Abwesenheit von Blutungen. Ein Ansprechen wird definiert als Thrombozytenzahl ≥ 30 x 10ˆ9/l und >2-facher Anstieg Thrombozytenzahl vom Ausgangswert sowie das Fehlen von Blutungen, beides bei 2 Gelegenheiten im Abstand von mehr als 7 Tagen gemessen 1.
Bei Kindern verläuft die idiopathische thrombozytopenische Purpura meist selbstlimitierend. Die Thrombozytenzahlen normalisieren sich nach ca. 2 bis 5 Wochen. Daher ist meistens ein abwartendes Vorgehen mit engmaschiger Kontrolle der Thrombozytenzahlen gerechtfertigt.
Zur Erstlinientherapie und Notfallbehandlung werden Kortikosteroide eingesetzt. Sie dichten die Kapillaren ab, reduzieren die Antiplättchen-Antikörpermenge und erhöhen die Plättchenproduktion. Kortikoidnebenwirkungen (siehe hier) sind zu berücksichtigen. Sinken die Thrombozytenzahlen unter 20.000 bis 30.000/ul, kommt eine Therapie mit Immunglobulinen oder intravenöses Anti-RhD-Immunglobulin infrage. Die Immunglobuline fangen die reaktiven Autoantikörper ab und verringern den Thrombozytenverlust. Sie wirken jedoch nicht dauerhaft. Anschließend kann mit Thrombopoietin-Rezeptor-Agonisten (TPO-RAs; Romiplostim oder Eltrombopag), Rituximab oder mit einer Splenektomie (einer operativen Milzentfernung) weiter behandelt werden 5.
Eine Splenektomie ist wirksam, weil die Milz Ort eines vermehrten Thrombozytenabbaus und gleichzeitig einer Antikörperbildung ist. Ein über 5 Jahre anhaltender Erfolg wurde bei 64 % der Patienten beobachtet. 6
Rituximab, ein monoklonaler Anti-CD20-Antikörper, unterdrückt die Bildung der Antiplättchen-Antikörper und darüber eine Wirksamkeit bei 40 bis 70 % der Patienten; es hat jedoch keine heilende Wirkung. 7
Thrombopoietin-Rezeptor-Agonisten (TPO-RA) fördern die Reifung der Megakaryozyten (Thrombozyten-Stammzellen) und steigern die Thrombozytenproduktion. Zur Verfügung stehen Romiplostim, Eltrombopag, Avatrombopag. Sie sollen einen Behandlungserfolg von etwa 80 % aufweisen. Als Risiko wird eine etwas erhöhte Thromboseneigung angegeben. 8
FcRn-Inhibitoren (Fc-Rezeptorenblocker) senken den Gesamt-IgG-Spiegel, ohne dessen Produktion zu beeinflussen oder die Spiegel anderer Immunglobulin-Isotypen zu verändern 9. Efgartigimod, ein Fc-Rezeptorblocker, führte in einer Studie zu einer raschen Senkung der Gesamt-IgG-Spiegel (bis zu 63,7 %) und einem klinisch relevanten Anstieg der Thrombozytenzahl. 46 % der Patienten erreichten unter Efgartigimod (vs. 25 % unter Placebo) mindestens zweimal eine Thrombozytenzahl von ≥ 50 × 10ˆ9 /l, und 38 % gegenüber 0 % erreichten ≥50 × 10ˆ9 /l an mindestens 10 Tagen hintereinander 10.
Hp-Assoziation: In Fällen einer idiopathischen thrombozytopenischen Purpura, in denen eine Helicobacter-Infektion des Magens festgestellt worden ist, konnte mehrfach eine deutliche Besserung oder Normalisierung der Thrombozytenzahl durch eine Helicobacter-Eradikation erzielt werden 11 3. In einer brasilianischen Studie wurde jedoch eine nur geringe, aber anhaltende Erhöhung der Thrombozytenzahl festgestellt, die zu einer Eradikationsbehandlung veranlassen sollte 12. Eine andere Studie bestätigt, dass eine H. pylori-Eradikation eine wirksame ITP-Behandlung für Patienten darstellt, die eine H. pylori-Gastritis aufweisen 13.
→ Auf facebook informieren wir Sie über Neues und Interessantes!
→ Verwalten Sie Ihre Laborwerte mit der Labor-App Blutwerte PRO – mit Lexikonfunktion.
Verweise
Weiteres
- J Blood Med. 2021 Jul 26;12:653-664. DOI: 10.2147/JBM.S259101.[↩][↩][↩]
- Semin Hematol. 2016 Apr;53 Suppl 1:S70-2[↩]
- Gut Liver. 2016 May 23;10(3):323-4[↩][↩]
- Rev Bras Hematol Hemoter. 2018 Jan-Mar;40(1):12-17. doi: 10.1016/j.bjhh.2017.09.005[↩]
- Semin Thromb Hemost. 2020 Apr;46(3):275-288. DOI: 10.1055/s-0039-1700512[↩]
- Br J Haematol. 2018;181(2):183–195. doi: 10.1111/bjh.15090[↩]
- Am J Hematol. 2019;94(12):1314–1324. doi: 10.1002/ajh.25632[↩]
- Ann Hematol. 2019;98(3):581–588. doi: 10.1007/s00277-018-3556-6[↩]
- Am J Hematol. 2024 Dec;99(12):2351-2366. doi: 10.1002/ajh.27487[↩]
- Am J Hematol. 2020 Feb;95(2):178-187. doi: 10.1002/ajh.25680[↩]
- Glob J Health Sci. 2015 Nov 3;8(7):52829. doi: 10.5539/gjhs.v8n7p35.[↩]
- Rev Bras Hematol Hemoter. 2018 Jan-Mar;40(1):12-17. DOI: 10.1016/j.bjhh.2017.09.005.[↩]
- Ann Hematol. 2022 Jul;101(7):1435-1445. doi: 10.1007/s00277-022-04782-2[↩]